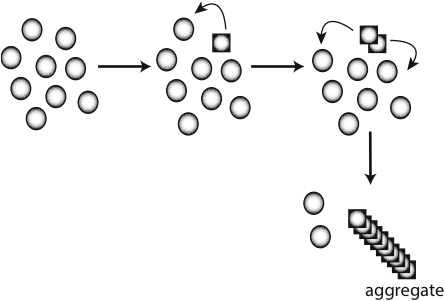
Figure 1. Early misfolding events associated with amyloid formation. A normally folded protein (circles) can undergo a rare conformation change (square) into a prion form. The altered conformation can further convert more normally folded versions of itself into the misfolded form. The aggregated form of the protein takes on beta rich structure and is called amyloid.
|
Protein misfolding is associated with many neurodegenerative (Alzheimer’s, Parkinson’s, Huntington’s, and Prion disease) and systemic (Amyloid Transthyretin and Light Chain Amyloidosis) diseases. In each of these diseases, a normal protein adopts an alternative but stable conformation that favors aggregation. Although there are genetic determinants that influence the formation of this alternative form, in the vast majority of patients the disease is caused by the sporadic misfolding of proteins. The focus of my lab is to understand the cellular mechanisms that underlie spontaneous protein misfolding associated with disease.
To begin to understand how these diseases work, our lab has focused on the formation of prions, which are associated with transmissible spongiform encephalopathies, a family of terminal neurological diseases that includes Creutzfeldt-Jakob disease in humans, and “Mad Cow” disease in cattle. In prion disease, a normally folded protein is converted to an alternate conformation that has the ability to further convert the normal protein into the misfolded infectious form (Fig 1). Although much work has focused on looking at infectivity and propagation of prions, much less is known about the initial phases of prion formation since the appearance rates in mammalian systems is approximately 1 in a million. Therefore, the study of a prion in baker’s yeast, [PSI+], has and will continue to be an important model to facilitate our understanding of prion appearance. |
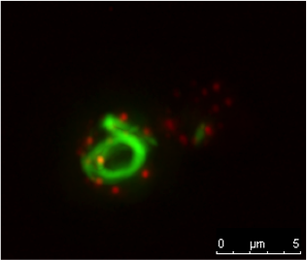
Newly formed Sup35-GFP rings with actin patches labeled by COF1-RFP. Photo by Doug Lyke
Yeast prions and actin networksTo uncover cellular pathways involved in prion formation, our previous work identified several yeast gene deletions that reduced the appearance of the [PSI+] prion. These gene deletions fell into two classes: one class of genes that are involved in the initial misfolding phases of prion appearance, and another class of genes that are important in later steps of prion formation. Interestingly, both classes of genes encode proteins that are involved in actin polymerization and appear to be required to maintain normal vacuolar morphology. Our current work focuses on both genetic approaches and protein biochemistry to investigate how actin networks and the vacuole play an important role in the multistep process of prion appearance.
|

Toxicity associated with expanded glutamine repeat of Human Huntingtin Exon 1 is suppressed by absence of the actin bundling protein Sac6p.
Using yeast to understand amyloid diseasesWe are using yeast as an in vivo test tube to determine how actin associated pathways impact amyloid formation of human proteins. Expanded glutamine tracts in exon 1 of the Huntington protein is associated with Human Huntington's disease, and causes toxicity in yeast. We have previously shown that introduction of these expanded glutamine alleles in some of our deletion mutants suppresses toxicity, whereas introduction in other gene deletion mutants enhances toxicity (click here for more info). Using both microscopy and genetic approaches, we are investigating how actin networks contribute to toxicity of these Human amyloid proteins. Our current focus is on identifying and characterizing cellular factors involved in misfolding of the mutant Transthyretin protein, associated with Amyloid Transthyretin (ATTR).
|
Recent Publications
Sharma J, Wisniewski BT, Paulson E, Obaoye JO, Merrill SJ, Manogaran AL. (2017) De novo [PSI+] prion formation involves multiple pathways to form infectious oligomers Sci Rep 7, 76 doi: 10.1038/s41598-017-00135-6.
Lyke, D. and Manogaran, AL. Spatial sequestration and oligomer remodeling during de novo [PSI+] formation. (in press) Prion.
Wisniewski, BT., Sharma, J., Legan, ER., Paulson, E., Merrill, SJ., and Manogaran, AL. (in press) Toxicity and Infectivity: insights from de novo prion formation. Curr Genet.
Goto pubmed for more publications. Click here.
Lyke, D. and Manogaran, AL. Spatial sequestration and oligomer remodeling during de novo [PSI+] formation. (in press) Prion.
Wisniewski, BT., Sharma, J., Legan, ER., Paulson, E., Merrill, SJ., and Manogaran, AL. (in press) Toxicity and Infectivity: insights from de novo prion formation. Curr Genet.
Goto pubmed for more publications. Click here.
